U każdego dziecka z rdzeniowym zanikiem mięśni (SMA) choroba przebiega inaczej. Wiek w momencie zachorowania, objawy, cechy SMA i stopień ciężkości choroby u poszczególnych dzieci mogą znacząco się różnić1.
Osoby przedstawione w tym materiale są prawdziwymi pacjentami, dlatego też uzyskano właściwą zgodę na używanie zdjęć od pacjentów oraz ich rodzin. Zdjęcia wyłącznie dla celów ilustracyjnych.
Udostępnij stronę
RDZENIOWY ZANIK MIĘŚNI U NIEMOWLĄT I DZIECI
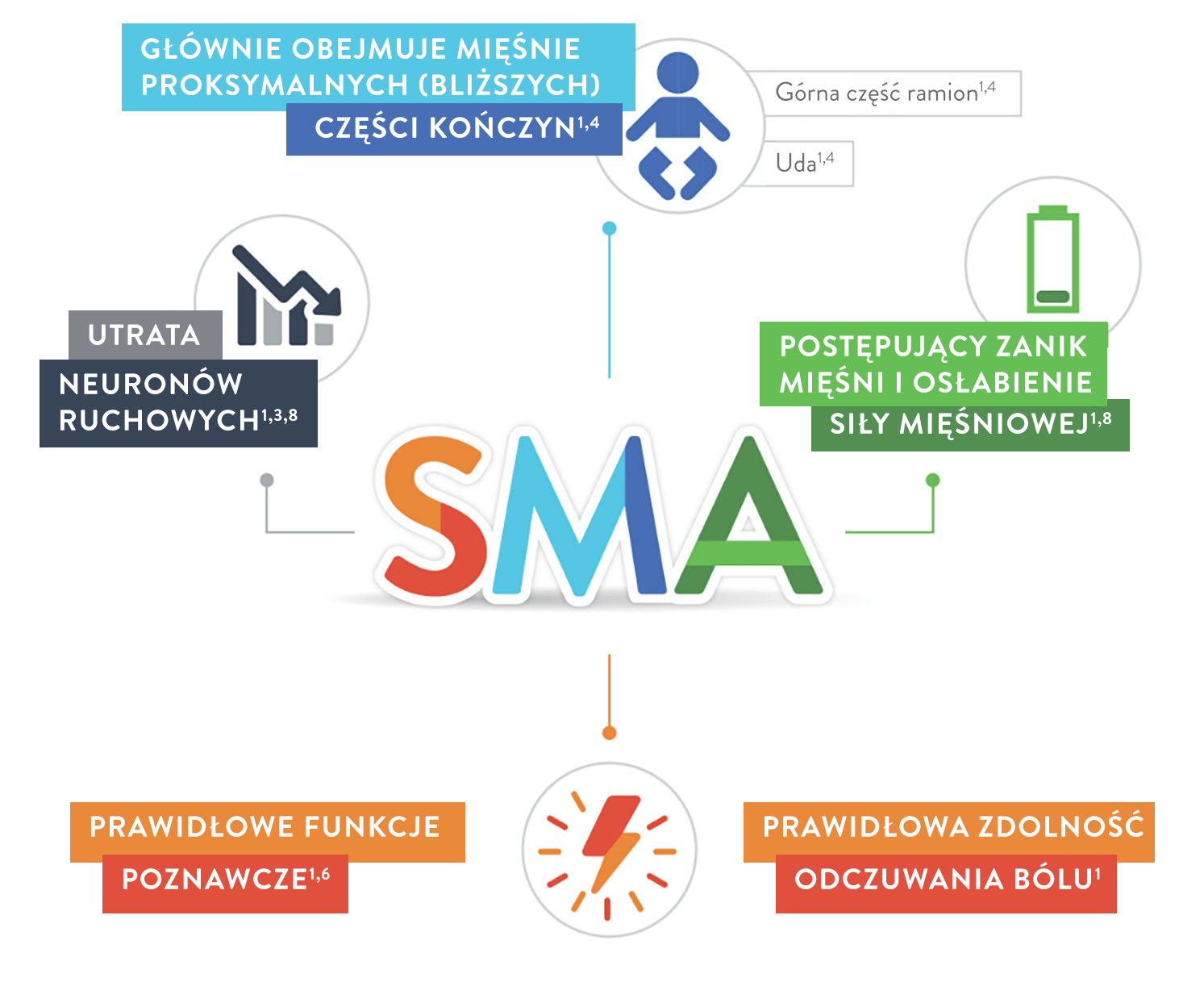
SMA jest rzadką, genetyczną chorobą nerwowo-mięśniową1,2
i wiodącą przyczyną śmiertelności z powodu zaburzeń genetycznych wśród niemowląt i małych dzieci3.
Dzieci mogą doświadczać postępującego osłabienia siły mięśni znajdujących się bliżej środka ciała, tak jak mięśnie barków, ud i miednicy. Mięśnie te umożliwiają wykonywanie takich czynności jak czworakowanie, chodzenie, siadanie i kontrolowanie ruchów głowy. U chorych można również obserwować zaburzenia oddychania i połykania4.
SMA nie zakłóca pracy neuronów odpowiedzialnych za procesy poznawcze służące do zdobywania wiedzy i większego rozumienia przez myślenie, doświadczenie i zmysły5,6.
Zgodnie z wynikami jednego badania dzieci i młodzież z SMA mają prawidłowy iloraz inteligencji (IQ)7.
Udostępnij teraz
U KAŻDEGO DZIECKA SMA PRZEBIEGA INACZEJ
Mogą one obejmować postępujące osłabienie mięśni, wiotkość i zaniki mięśniowe. Osłabienie mięśniowe występuje zazwyczaj symetrycznie po obu stronach ciała9.
Każde dziecko może inaczej doświadczać objawów choroby, a SMA dzielimy na różne typy w zależności od wieku w momencie zachorowania i zdolności czynnościowych pacjenta. Choroba danego typu może mieć również różne nasilenie, jednak nawet u 25% osób z rozpoznaniem SMA nie można jednoznacznie określić typu zaburzenia10.
Udostępnij teraz
Udostępnij teraz
Osoby przedstawione w tym materiale są prawdziwymi pacjentami, dlatego też uzyskano właściwą zgodę na używanie zdjęć od pacjentów oraz ich rodzin. Zdjęcia wyłącznie dla celów ilustracyjnych.
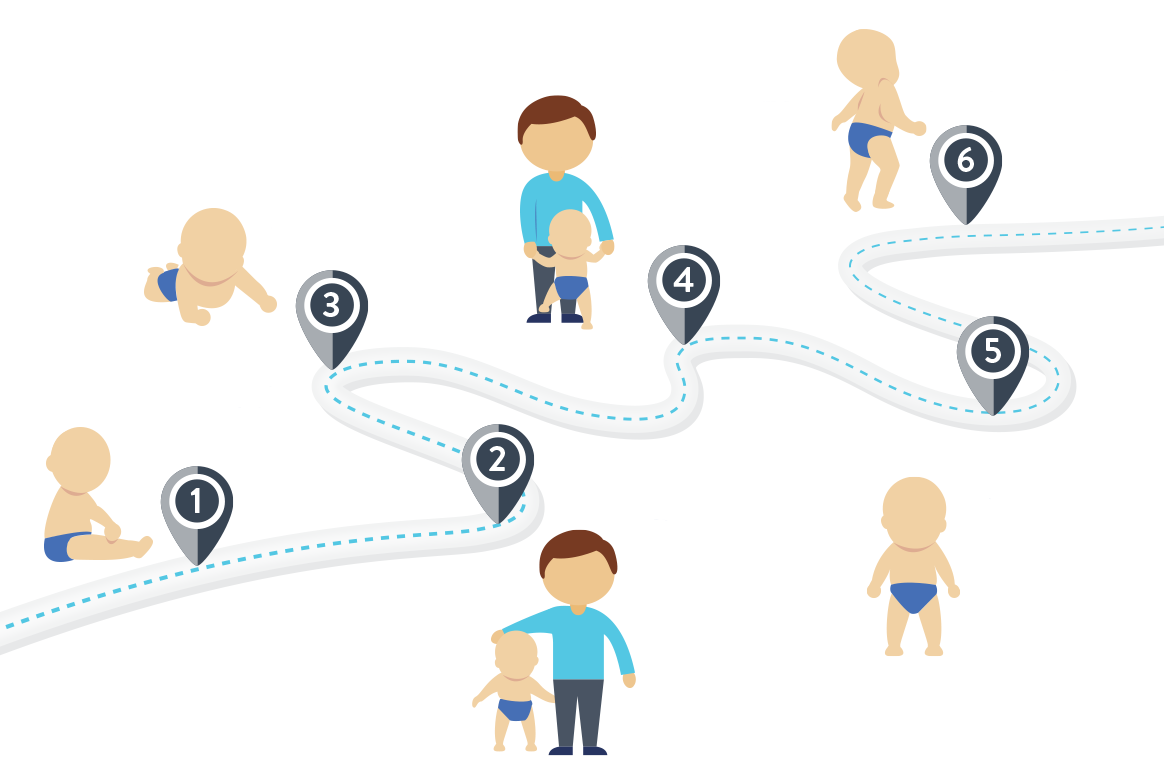
NIEOSIĄGNIĘCIE NIEKTÓRYCH KAMIENI MILOWYCH ROZWOJU MOŻE BYĆ PIERWSZĄ OZNAKĄ SMA
Podejrzenie rdzeniowego zaniku mięśni często jako pierwszy wysuwa rodzic, który zauważa, że jego dziecko nie osiąga niektórych kamieni milowych rozwoju. Rodzice mogą zauważyć, że dziecko nie osiąga typowych kamieni milowych rozwoju fizycznego przypisanych dla wieku, jak umiejętność trzymania za rękę, obracanie się lub siadanie bez podparcia.
U chorych mogą również pojawić się trudności w połykaniu lub karmieniu. Dzieci mogą utracić zdolność bezpiecznego połykania bez przypadków dławienia lub aspiracji pokarmów do dróg oddechowych5,11.
Chociaż wszystkie dzieci rozwijają się w własnym tempie, Światowa Organizacja Zdrowia (WHO), w ramach badania Multicentre Growth Reference Study (MGRS), opracowała ogólne wytyczne dotyczące osiągania kamieni milowych rozwoju ruchowego12.
Udostępnij teraz
SIEDZI BEZ PODPARCIA
4–9 MIESIĘCY
CZWORAKUJE
5,5–13,5 MIESIĄCA
CHODZI Z PODPARCIEM
6-14 MIESIĘCY
CHODZI SAMODZIELNIE
8-18 MIESIĘCY
STOI SAMODZIELNIE
7-16 MIESIĘCY
STOI Z PODPARCIEM
5-11 MIESIĘCY
Share now
Na podstawie: WHO Multicentre Growth Reference Study Group, 200613
WYTYCZNE OSIĄGANIA KAMIENI MILOWYCH ROZWOJU RUCHOWEGO12
GŁÓWNE KAMIENIE MILOWE ROZWOJU RUCHOWEGO
KRYTERIA SPRAWNOŚCI
1.Siedzenie bez podparcia
Dziecko siedzi prosto z uniesioną głową przez ≥10 sekund.
Nie używa ramion ani rąk do utrzymania równowagi lub podparcia.
2.Czworakowanie
Dziecko porusza się do przodu lub do tyłu na rękach i kolanach.
Brzuch nie dotyka powierzchni podparcia.
Nieprzerwane i następujące po sobie ruchy (≥3 z rzędu).
3.Stanie z podparciem
Dziecko stoi na obu nogach w pozycji wyprostowanej, trzymając się stabilnego przedmiotu, takiego jak meble.
Dziecko stoi, trzymając się przez ≥10 sekund.
4.Chodzenie z podparciem
Dziecko stoi w pozycji wyprostowanej z wyprostowanymi plecami.
Wykonuje kroki w bok lub do przodu, trzymając się stabilnego przedmiotu jedną lub obiema rękami.
Jedna noga porusza się do przodu, podczas gdy druga podtrzymuje ciężar ciała.
Dziecko wykonuje ≥5 kroków.
5.Stanie bez podparcia
Dziecko stoi w pozycji wyprostowanej na obu stopach (ale nie na palcach) z wyprostowanymi plecami.
Nogi utrzymują 100% masy ciała, bez przytrzymywania, przez ≥10 sekund.
6.Chodzenie bez podparcia
Dziecko wykonuje ≥5 kroków bez podparcia z wyprostowanymi plecami.
Jedna noga porusza się, podczas gdy druga podtrzymuje większość ciężaru ciała.
Nie ma kontaktu z osobą lub przedmiotem.
Udostępnij teraz
Osoby przedstawione w tym materiale są prawdziwymi pacjentami, dlatego też uzyskano właściwą zgodę na używanie zdjęć od pacjentów oraz ich rodzin. Zdjęcia wyłącznie dla celów ilustracyjnych.
CHARAKTERYSTYKA SMA U NIEMOWLĄT I DZIECI
Dowiedz się więcej na temat cech i różnych typów SMA w zależności od wieku w momencie zachorowania:
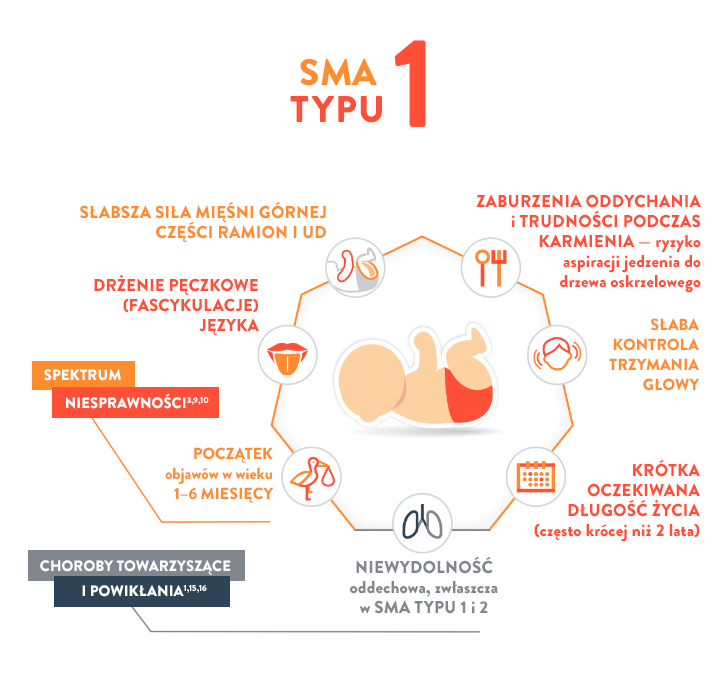
Potrafi utrzymać głowę i zaczyna się podnosić, leżąc na brzuchu; wykonuje płynniejsze ruchy rękami i nogami.
4 miesiąc życia:
Utrzymuje głowę stabilnie bez podparcia; naciska na nogi, gdy stopy znajdują się na twardej powierzchni; może się obrócić z brzucha na plecy; może trzymać i kołysać wiszącymi zabawkami; podnosi ręce do ust; leżąc na brzuchu, naciska na łokcie.
6 miesiąc życia:
Obraca się w obu kierunkach (z brzucha na plecy i z pleców na brzuch); zaczyna siedzieć bez podparcia; stojąc, opiera ciężar ciała na nogach i może się podskakiwać; kołysze się w przód i w tył, czasami czołga się do tyłu przed pójściem do przodu.
Najważniejszy, osiągnięty kamień milowy rozwoju ruchowego:
Nie potrafią siedzieć („niesiedzące”)
Oczekiwana długość życia:
≤2 lat14
Typ SMA
Typ 1 (zwany również chorobą Werdniga-Hoffmanna)
CECHY14,17,18:
Słaba kontrola trzymania głowy
Słaby odruch kaszlowy
Słaby płacz
Postępujące osłabienie mięśni odpowiedzialnych za żucie i połykanie
Słabe napięcie mięśniowe
Nogi ułożone jak żaba w pozycji leżącej
Ciężkie osłabienie siły mięśniowej po obu stronach ciała
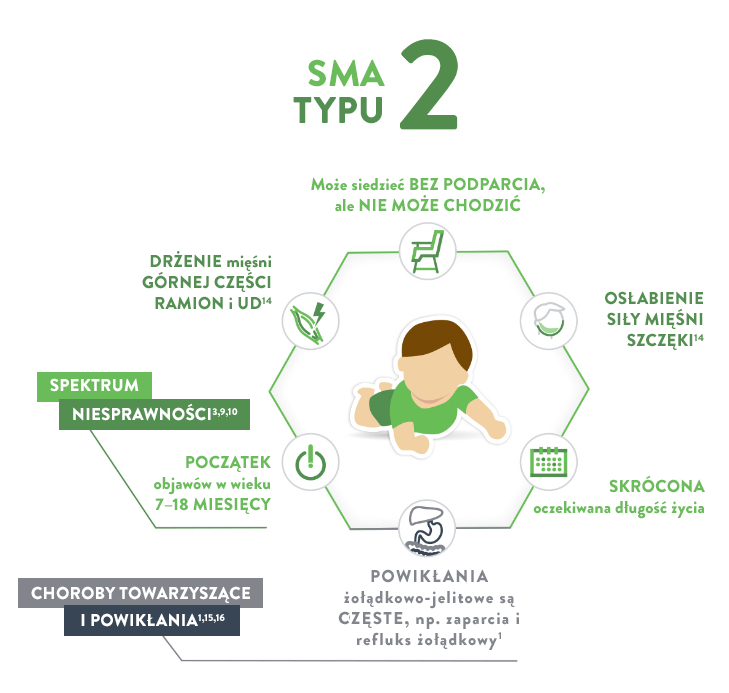
Stoi, przytrzymując się przedmiotów; potrafi przejść do pozycji siedzącej; siedzi bez podparcia; odpycha się, żeby stanąć; czworakuje.
1 rok:
Samodzielnie przechodzi do pozycji siedzącej; odpycha się, żeby stanąć; chodzi, trzymając się mebli (bokiem); potrafi wykonać kilka kroków bez podparcia; potrafi stać bez podparcia.
Najważniejszy, osiągnięty kamień milowy rozwoju ruchowego:
Potrafią siedzieć bez podparcia („siedzący”)
Oczekiwana długość życia
> 2 lat14
70% dożywa wieku 25 lat
Typ SMA
Typ 2 (zwany również chorobą Dubowitza)
CECHY17, 18:
Osłabienie mięśni
Mogą występować problemy z połykaniem, odruchem kaszlowym i oddychaniem, ale zazwyczaj występują rzadziej
Bóle mięśni i sztywność stawów
U dzieci mogą rozwijać się takie problemy, jak skolioza (boczne skrzywienie kręgosłupa), która może wymagać stosowania ortez lub leczenia chirurgicznego
Udostępnij teraz
Najważniejszy, osiągnięty kamień milowy rozwoju ruchowego:
Potrafią samodzielnie chodzić („chodzący” – chociaż mogą stopniowo tracić tę zdolność)
Oczekiwana długość życia
Prawidłowa14
Typ SMA
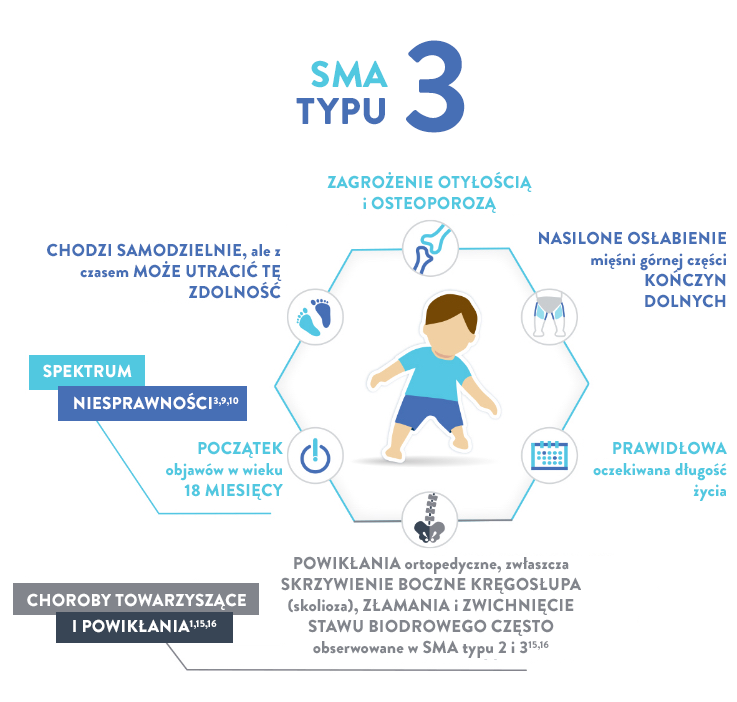
Typ 3 (zwany również chorobą Kugelberga-Welandera)
CECHY17,18:
Skolioza (boczne skrzywienie kręgosłupa)
Zaburzenia żucia i połykania
Mięśnie nóg są zazwyczaj w większym stopniu dotknięte procesem chorobowym niż mięśni rąk
Bóle mięśni
Objawy wynikające z nadmiernego używania stawów
Udostępnij teraz
Najważniejszy, osiągnięty kamień milowy rozwoju ruchowego
Wszystkie
Oczekiwana długość życia
Prawidłowa14
Typ SMA
Typ 4
CECHY17,18:
Objawy fizyczne są podobne jak w rdzeniowym zaniku mięśni o początku w wieku młodzieńczym z postępującym osłabieniem drżeniami i skurczami mięśni pojawiającymi się w późnym wieku młodzieńczym lub wczesnym wieku dorosłym.
Udostępnij teraz
Udostępnij teraz
Udostępnij teraz
POSŁUCHAJ OPINII RODZINY LAMONTÓW
„Czas do ustalenie rozpoznania jest bardzo ważny, ponieważ pomaga rodzinom uzyskać wsparcie, którego potrzebują.”
– mama Lilah
Obejrzyj film wideo terazvideoWrapper1
Informacje na temat rozwoju umiejętności dziecka z zakresu motoryki dużej podawane przez rodziców są zazwyczaj wiarygodne. Przekazywane lekarzowi informacji na temat potencjalnych opóźnień w rozwoju ruchowym może pomóc określić odpowiednią strategię opieki19,20
Osoby przedstawione w tym materiale są prawdziwymi pacjentami, dlatego też uzyskano właściwą zgodę na używanie zdjęć od pacjentów oraz ich rodzin. Zdjęcia wyłącznie dla celów ilustracyjnych.
Piśmiennictwo
1. Wang CH, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-1049.
3. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
4. Finkel R, et al. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7-9. November 2014, Heemskerk, The Netherlands. Neuromuscul Disord. 2015;25(7):593-602.
5. Qian Y., McGraw S., Henne J., Jarecki J., Hobby K., Yeh W.S. Understanding the experiences and needs of individuals with Spinal Muscular Atrophy and their parents: A qualitative study. BMC Neurol. 2015;15:1–12. doi: 10.1186/s12883-015-0473-3.
7. Von Gontard et al. Intelligence and cognitive function in children and adolescents with spinal muscular atrophy. Neuromuscul Disord. 2002. Feb;12(2):130-6.
11. Centers for Disease Control and Prevention. Developmental milestones. Available at: http://www.cdc.gov/ncbddd/actearly/milestones/. Updated January 21, 2016. Accessed April 27, 2016.
12. Wijnhoven TMA, de Onis M, Onyango AE, et al; for the WHO Multicentre Growth Reference Study Group. Assessment of gross motor development in the WHO Multricentre Growth Reference Study. Food Nutr Bull. 2004;25(1 suppl 1):S37-S45.
14. Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol. 2012;46(1):1-12.
15. Haaker G, Fujak A. Proximal spinal muscular atrophy: current orthopedic perspective. Appl Clin Genet 2013;6:113-120.
16. Darras BT. Spinal muscular atrophies. Paediatr Clin North Am 2015;62(3):743-766. DOI: 10.1016/j.pcl.2015.03.010.
17. Mercuri E, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscl Disord. 2018;28(2):103-115.
18. Prior TW, Russman BS. Spinal muscular atrophy. NCBI Bookshelf Website. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1352/. Updated November 14, 2013. Accessed April 15, 2016.
19. Noritz GH, Murphy NA; and Neuromuscular Screening Expert Panel. Motor delays: early identification and evaluation. Pediatrics. 2013;131(6):e2016-e2027.
20. Lawton S, Hickerton C, Archibald AD, McClaren BJ, Metcalfe SA. A mixed methods exploration of families’ experiences of the diagnosis of childhood spinal muscular atrophy. Eur J Hum Genet. 2015;23(5):575-580.